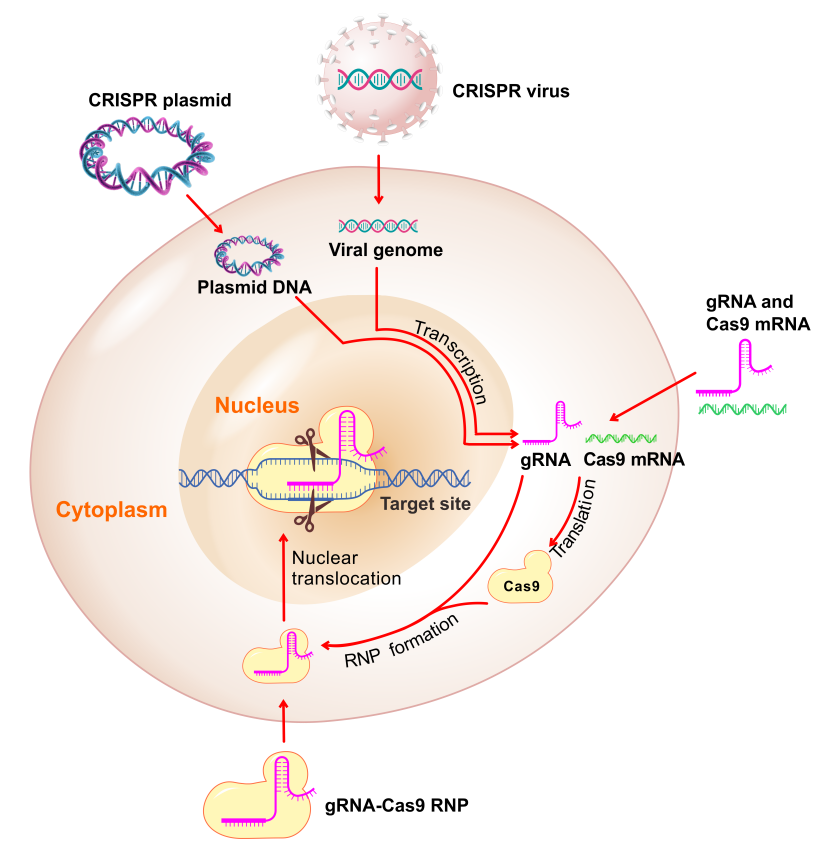
CRISPR系统和TALEN系统均能用于在体外细胞和模式生物这种进行基因编辑。以下是这两种系统的特性对比:
基本原理
CRISPR
CRISPR系统使用位点特异的向导RNA (gRNA) 将Cas9核酸酶引导至其基因组中的目标位点以产生DNA切割。靶序列的长度通常约为20 bp。若靶序列中包含一些错配位点仍可能被识别和切割。
了解CRISPR载体的更多信息
TALEN
TALEN系统使用的是一对嵌合蛋白,每个嵌合蛋白包含一个融合到Fokl核酸酶结构域的且作为TAL效应子的DNA结合结构域(识别特定DNA序列)。这对蛋白被设计成与基因组中的一对靶位点结合,每个靶位点长约18 bp,且二者相隔一个14-20 bp的间隔区。在与 DNA 结合后,这对蛋白上的Fokl核酸酶结构域发生二聚化,然后导致两个靶位点之间的间隔区内的序列发生切割。
效率
CRISPR和TALEN系统均在基因编辑上均表现出良好的效率,这也具体取决于应用的物种和细胞类型。一般来说,CRISPR系统的组分进入细胞后引发的DNA切割比TALEM系统更高效。
脱靶效应
CRISPR系统的gRNA靶向约20 bp的DNA序列,而TALEN系统需要约36 bp的靶序列。此外,Cas9/gRNA 复合物对靶序列中碱基错配(最多5 bp错配)的容忍度高于 TALEN。因此,TALEN介导的DNA切割比CRISPR具有更好的特异性,也不太可能在基因组中发生脱靶性的切割。相比之下,已有报道表明CRISPR系统在体外细胞系中可产生脱靶效应,而对CRISPR敲除小鼠的分析则表明体内实验时脱靶频率较低。最新的CRISPR系统的显着增强了CRISPR的特异性。通过使用双gRNA和Cas9切口酶(仅包含一个具有催化活性的核酸酶结构域的Cas9突变体,如Cas9(D10A)和Cas9_H840A),在靶向区域附近产生两个单链DNA切口,从而导致DSB发生在可修复的靶向区域内。在这种设计中由于双gRNA可将靶向序列的长度扩展至约40 bp,因此最大限度地减少了脱靶效应。
靶位点要求
TALEN系统可以针对基因组中的几乎任何位置进行设计。相比之下,CRISPR系统中的靶位点选择受限于PAM序列(通常为NGG)的要求,该序列位于gRNA靶向的DNA序列的3'末端。CRISPR系统的这种特性这并不会阻碍基因敲除,因为对靶基因中的任何地方切割都是有效的基因编辑,但可能难以对基因的特定位置进行切割或实现位点的特异性突变。为了通过将CRISPR系统应用于可精确编辑特定基因组位点,可以将包含所需编辑序列的同源重组供体载体或长寡核苷酸与靶位点的上游和下游同源臂一同递送至细胞中,以指导靶位点发生HDR介导的DNA修复。
点击设计您的基因打靶供体载体
技术难度
在基因编辑实验中,使用CRISPR系统在几个方面表现出对比TALEN具有更低的技术难道。 首先,对于载体构建,CRISPR 系统只需要合成一个短的gRNA,因为Cas9/gRNA 复合物的靶向依赖于机制简单的RNA/DNA杂交,而TALEN 系统需要根据每个独特的蛋白-DNA相互作用重新设计TAL的DNA结合域。与TALEN系统相比,gRNA显然是更便宜、更容易设计和构建的,而且TALEN系统打靶每个位点总是需要两个载体。其次,对于一些应用,例如注射小鼠胚胎,Cas9蛋白和gRNA可以通过直接注射来高效地递送,但TALEN系统不能。最后,CRISPR系统在基因筛选实验中的应用非常广泛,表达数千种不同gRNA的CRISPR筛选文库可以很容易地以高通量方式构建。